Part 2 : Going further using bedtools and awk¶
Genomic coordinates¶
Working with genomic data frequently implies to deal with biological sequences. As these sequences (genes, exons, ...) are related to genomes, handling genomic coordinates is typically required. A basic task in genomic data analysis is to compare different sets of genomic features (transcripts, promoter regions, polymorphisms, conserved elements,...). One question could be for instance “which SNPs (Single Nucleotide Polymorphisms) are associated with a disease of interest and fall into exonic regions”. Such questions require dedicated tools to ease data analysis.
Bedtools has been developed to compare sets of genomic features and is now becoming a standard Linux tool for people working in the field. Bedtools largely relies on the BED file format (although it may also operate with GFF/GTF, VCF, and SAM/BAM files).
The Bed file format is a very simple way to store information related to genomic features. A typical file in BED format will contain the following columns (the 3 first columns are mandatory):
- chrom - The name of the chromosome (e.g. chr3, chrY, chr2...).
- chromStart - The starting position of the feature in the chromosome.
- chromEnd - The ending position of the feature in the chromosome.
- name - A name for the feature (e.g. gene name...).
- score - A score between 0 and 1000.
- strand - Defines the strand - either ‘+’ or ‘-‘.
Which fraction of the human genome is covered by exons ?¶
In the section below we will try to answer the following question: “Which fraction of the genome is covered by exons ?”.
Downloading genomic coordinates of exons.¶
Download the following
filewhich contains the coordinates of the exons of the RefSeq transcripts version hg38.Using the
mvcommand, move the Downloaded file into the directory Bioinfo_WS1718/data.- Look at the file using
lessto see the format. Is this a bed format ? - How many exons do we have in total ?
- Tricky one : can you find again the transcript which has the largest number of exons ?
- Look at the file using
1 2 3 4 5 6 7 | # this is a genuine bed format
wc -l hg38_exons.bed
cut -f4 hg38_exons.bed | cut -d ";" -f1 | uniq -c | sort -nr
# first command extracts the column with the transcript name
# second extracts from this only the string which is left of the ";"
# third counts how often this transcript name occurs
# 4th sorts the results.
|
Merging overlapping regions¶
We will use a “genomic toolbox” called Bedtools, which offers a large number of utilities to manage genomic datasets.
To answer our simple question, we must first keep in mind that several exons may overlap, due to various phenomena (alternative splicing, multiple promoters or terminators, mutually overlapping genes). To avoid counting several times the same region, our first task will thus be to merge these overlapping regions. The bedtools merge command from the Bedtools suite combines overlapping features in an interval file into a single feature which spans all of the combined features. The image below illustrate this.
Warning
Beware: bedtools merge requires the genomic coordinates to be sorted (see below).
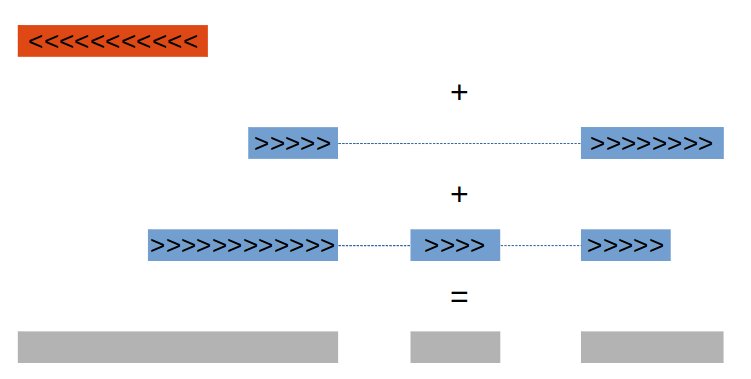
- Get some help about the
bedtools sortcommand using the -h (help) argument. - Use the
bedtools sortcommand to sort exons by coordinates and store the results in hg38_exons_sorted.bed. - Get some help about the
bedtools mergecommand using the -h (help) argument. - Use
bedtools mergewith hg38_exons_sorted.bed as input to combine overlapping exons into single features and store the results into a file named mergedExons.bed. - Have a look at the first lines with the
headcommand. - The length of one genomic feature can simply be obtained by computing column_3-column_2. Use a awk command to compute the sum of the length of all features (check the pdf of the lecture on bash and awk
Vorlesung Bash). - Compute the total length of the genome using
this file. - Now, what is the fraction of the genome that is covered by exons or genes ?
1 2 3 4 5 6 7 8 9 10 | bedtools sort -i hg38_exons.bed > hg38_exons_sorted.bed
bedtools merge -i hg38_exons_sorted.bed > hg38_exons_merged.bed
# total length of all exons
awk 'BEGIN {l=0} {l=l+$3-$2+1} END {print l}' hg38_exons_merged.bed
# total length of the genome
awk 'BEGIN {l=0} {l=l+$2} END {print l}' chromInfo.txt
# then simply divide the first number by the second using a calculator ...
|